Deep Learning framework for virtual screening in drug discovery
Tech, data & sensors:
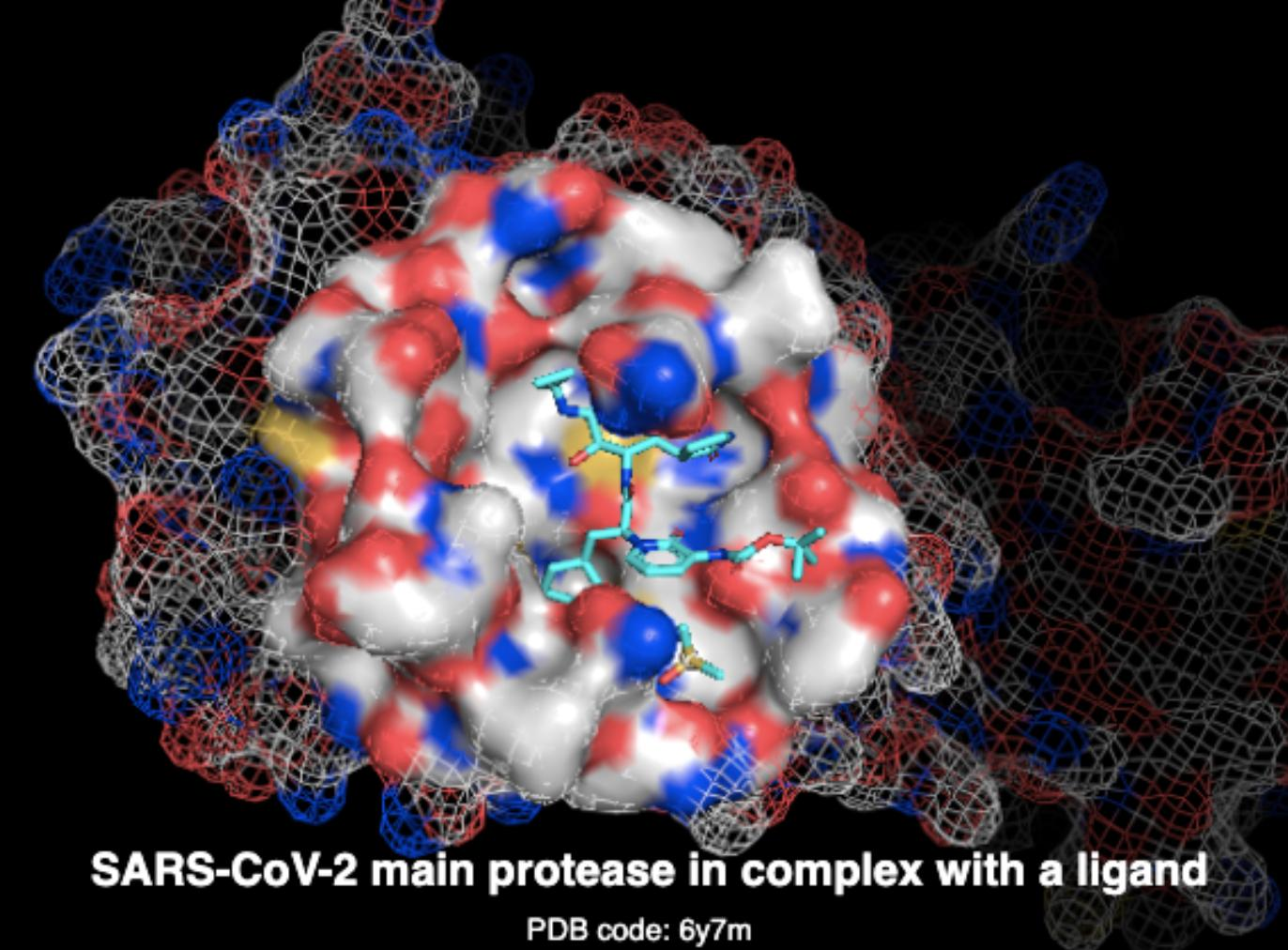
3D surface, graphs, protein and ligand molecules, geometric deep learning
Challenge
In the pharmaceutical industry, the average new drug development is approximately $2.6 billion, while the success rates are typically below 10%. One of the first phases of drug discovery for a chosen target concerns high-throughput screening. Machine learning has the potential to substantially accelerate the drug discovery lifecycle by performing virtual screening of large databases of candidate drug molecules against a specific target (e.g., a protein) associated with some known disease.Solution
We developed a deep learning solution based on state-of-the-art geometric deep learning technology for 3D protein-surface [Gainza 2020] and molecular-graph modelling for the ligand [Hu 2019] and subsequently fused the two modalities to predict a binding affinity score. We evaluated the above solution in various benchmark datasets with impressive results.Results
We virtually screened billions of ligands as part of the JEDI worldwide Grand Challenge. Our team currently is among the top-20 finalist teams by which our selected molecules will be experimentally validated in the laboratory for potential drug-treatment for COVID19.Bibliography
- Gainza, P., Sverrisson, F., Monti, F., Rodolà, E., Boscaini, D., Bronstein, M. M., & Correia, B. E. (2020). Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nature Methods, 17(2), 184–192.
- Hu, W., Liu, B., Gomes, J., Zitnik, M., Liang, P., Pande, V., & Leskovec, J. (2019). Strategies for Pre-training Graph Neural Networks. 1, 1–5.